
Genetic variant carried by 60% of Europeans predisposes people to slimness CNIO_Cancer
For mouse tissues and primary cells, RNA was extracted using TRIzol together with RNeasy Mini Kit or Direct-zol RNA Miniprep . Reverse transcription was performed using SuperScript IV VILO Master Mix . Quantitative real-time PCR was run in triplicates using GoTaq qPCR Master Mix in a QuantStudio 6 Flex Real-Time PCR System thermocycler . The 2as reference gene.
A final multifactorial model to predict BMI was validated through bootstrap, using 2000 bootstrap samples, which allowed to correct the performance statistics by optimism in order to assess its predictive power and possible overfit. This final model was obtained by sequentially adding SNP and gene expression variables to a basal model with only sex and age as predictor variables. A new variable was accepted if it was statistically significant and in addition produced a new model with an increased optimism-corrected, in order to prevent overfit. In the final model, the relative importance for the different predictor variables was estimated from the χAll tests were bilateral, with a significance level of 0.05.
United Kingdom Latest News, United Kingdom Headlines
Similar News:You can also read news stories similar to this one that we have collected from other news sources.
 scTAM-seq enables targeted high-confidence analysis of DNA methylation in single cells - Genome BiologySingle-cell DNA methylation profiling currently suffers from excessive noise and/or limited cellular throughput. We developed scTAM-seq, a targeted bisulfite-free method for profiling up to 650 CpGs in up to 10,000 cells per experiment, with a dropout rate as low as 7%. We demonstrate that scTAM-seq can resolve DNA methylation dynamics across B-cell differentiation in blood and bone marrow, identifying intermediate differentiation states that were previously masked. scTAM-seq additionally queries surface-protein expression, thus enabling integration of single-cell DNA methylation information with cell atlas data. In summary, scTAM-seq is a high-throughput, high-confidence method for analyzing DNA methylation at single-CpG resolution across thousands of single cells.
scTAM-seq enables targeted high-confidence analysis of DNA methylation in single cells - Genome BiologySingle-cell DNA methylation profiling currently suffers from excessive noise and/or limited cellular throughput. We developed scTAM-seq, a targeted bisulfite-free method for profiling up to 650 CpGs in up to 10,000 cells per experiment, with a dropout rate as low as 7%. We demonstrate that scTAM-seq can resolve DNA methylation dynamics across B-cell differentiation in blood and bone marrow, identifying intermediate differentiation states that were previously masked. scTAM-seq additionally queries surface-protein expression, thus enabling integration of single-cell DNA methylation information with cell atlas data. In summary, scTAM-seq is a high-throughput, high-confidence method for analyzing DNA methylation at single-CpG resolution across thousands of single cells.
Read more »
 Personalising whole genome sequencing doubles diagnosis of rare diseasesTailoring the analysis of whole genome sequencing to individual patients could double the diagnostic rates of rare diseases, finds a new study led by UCL researchers.
Personalising whole genome sequencing doubles diagnosis of rare diseasesTailoring the analysis of whole genome sequencing to individual patients could double the diagnostic rates of rare diseases, finds a new study led by UCL researchers.
Read more »
 Viral whole genome study in the United States identifies ORF-disrupting mutations in monkeypox virusResearchers used whole genome sequencing to evaluate monkeypox virus samples from Washington and Ohio states in the United States and identified 25 samples with open reading frame (ORF)-disrupting mutations.
Viral whole genome study in the United States identifies ORF-disrupting mutations in monkeypox virusResearchers used whole genome sequencing to evaluate monkeypox virus samples from Washington and Ohio states in the United States and identified 25 samples with open reading frame (ORF)-disrupting mutations.
Read more »
 Phylodynamic analysis of the monkeypox virus genome suggests low genetic variation and evolutionary ratesResearchers explored the genetic variability among the monkeypox virus genomes of Clade IIb lineage B.1 using a phylodynamic and genetic survey to confirm the epidemic cluster in the clade and understand the evolution of the virus.
Phylodynamic analysis of the monkeypox virus genome suggests low genetic variation and evolutionary ratesResearchers explored the genetic variability among the monkeypox virus genomes of Clade IIb lineage B.1 using a phylodynamic and genetic survey to confirm the epidemic cluster in the clade and understand the evolution of the virus.
Read more »
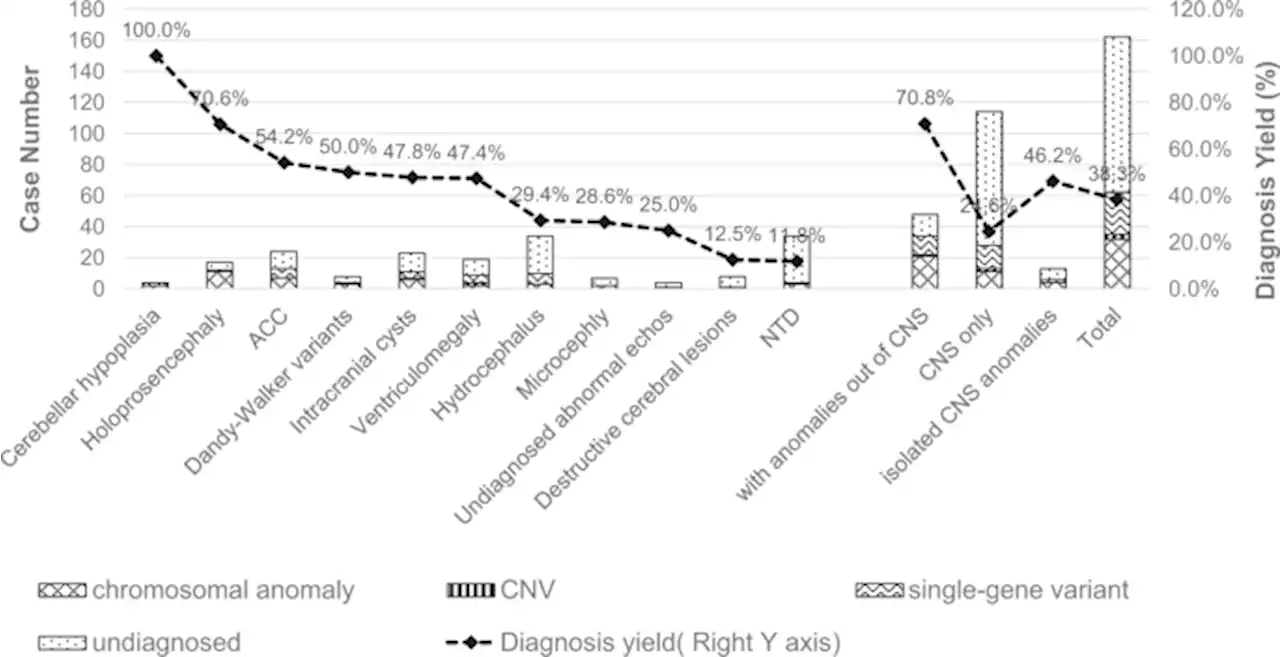 Genomic architecture of fetal central nervous system anomalies using whole-genome sequencing - npj Genomic Medicinenpj Genomic Medicine - Genomic architecture of fetal central nervous system anomalies using whole-genome sequencing
Genomic architecture of fetal central nervous system anomalies using whole-genome sequencing - npj Genomic Medicinenpj Genomic Medicine - Genomic architecture of fetal central nervous system anomalies using whole-genome sequencing
Read more »
 Mendelian randomization analysis of factors related to ovulation and reproductive function and endometrial cancer risk - BMC MedicineBackground Observational epidemiological studies suggest a link between several factors related to ovulation and reproductive function and endometrial cancer (EC) risk; however, it is not clear whether these relationships are causal, and whether the risk factors act independently of each other. The aim of this study was to investigate putative causal relationships between the number of live births, age at last live birth, and years ovulating and EC risk. Methods We conducted a series of observational analyses to investigate various risk factors and EC risk in the UK Biobank (UKBB). Additionally, multivariate analysis was performed to elucidate the relationship between the number of live births, age at last live birth, and years ovulating and other related factors such as age at natural menopause, age at menarche, and body mass index (BMI). Secondly, we used Mendelian randomization (MR) to assess if these observed relationships were causal. Genome-wide significant single nucleotide polymorphisms (SNPs) were extracted from previous studies of woman’s number of live births, age at menopause and menarche, and BMI. We conducted a genome-wide association analysis using the UKBB to identify SNPs associated with years ovulating, years using the contraceptive pill, and age at last live birth. Results We found evidence for a causal effect of the number of live births (inverse variance weighted (IVW) odds ratio (OR): 0.537, p = 0.006), the number of years ovulating (IVW OR: 1.051, p = 0.014), in addition to the known risk factors BMI, age at menarche, and age at menopause on EC risk in the univariate MR analyses. Due to the close relationships between these factors, we followed up with multivariable MR (MVMR) analysis. Results from the MVMR analysis showed that number of live births had a causal effect on EC risk (OR: 0.783, p=0.036) independent of BMI, age at menarche and age at menopause. Conclusions MVMR analysis showed that the number of live births causally reduced the ri
Mendelian randomization analysis of factors related to ovulation and reproductive function and endometrial cancer risk - BMC MedicineBackground Observational epidemiological studies suggest a link between several factors related to ovulation and reproductive function and endometrial cancer (EC) risk; however, it is not clear whether these relationships are causal, and whether the risk factors act independently of each other. The aim of this study was to investigate putative causal relationships between the number of live births, age at last live birth, and years ovulating and EC risk. Methods We conducted a series of observational analyses to investigate various risk factors and EC risk in the UK Biobank (UKBB). Additionally, multivariate analysis was performed to elucidate the relationship between the number of live births, age at last live birth, and years ovulating and other related factors such as age at natural menopause, age at menarche, and body mass index (BMI). Secondly, we used Mendelian randomization (MR) to assess if these observed relationships were causal. Genome-wide significant single nucleotide polymorphisms (SNPs) were extracted from previous studies of woman’s number of live births, age at menopause and menarche, and BMI. We conducted a genome-wide association analysis using the UKBB to identify SNPs associated with years ovulating, years using the contraceptive pill, and age at last live birth. Results We found evidence for a causal effect of the number of live births (inverse variance weighted (IVW) odds ratio (OR): 0.537, p = 0.006), the number of years ovulating (IVW OR: 1.051, p = 0.014), in addition to the known risk factors BMI, age at menarche, and age at menopause on EC risk in the univariate MR analyses. Due to the close relationships between these factors, we followed up with multivariable MR (MVMR) analysis. Results from the MVMR analysis showed that number of live births had a causal effect on EC risk (OR: 0.783, p=0.036) independent of BMI, age at menarche and age at menopause. Conclusions MVMR analysis showed that the number of live births causally reduced the ri
Read more »